
 ANSI/HL7 V3 ICSRP2, R2-2012 HL7 Version 3 Standard: Pharmacovigilance - Individual Case Safety Report, Part 2: Human Pharmaceutical Reporting Requirements for ICSR, R2 (revise and partition ANSI/HL7 V3 RRCS, R1-2005) 1/31/2012 |
Content Last Edited: 2012-02-20T17:13:56
3.2 Storyboards
3.3 Application Roles
3.4 Trigger Events
3.5 Refined Message Information Models
3.6 Hierarchical Message Descriptions
3.7 Interactions
Foreword
This standard is published as a SDO Joint Harmonization International Standard ISO 27953 and was prepared by Technical Committee ISO/TC 215, Health Informatics, CEN TC 251 Health Informatics and HL7 Patient Safety Workgroup.
ISO (the International Organization for Standardization) is a worldwide federation of national standards bodies (ISO member bodies). The work of preparing International Standards is normally carried out through ISO technical committees. Each member body interested in a subject for which a technical committee has been established has the right to be represented on that committee. International organizations, governmental and non-governmental, in liaison with ISO, also take part in the work. ISO collaborates closely with the International Electrotechnical Commission (IEC) on all matters of electrotechnical standardization.
International Standards are drafted in accordance with the rules given in the ISO/IEC Directives, Part 2. The main task of technical committees is to prepare International Standards. Draft International Standards adopted by the technical committees are circulated to the member bodies for voting. Publication as an International Standard requires approval by at least 75 % of the member bodies casting a vote.
Attention is drawn to the possibility that some of the elements of this document may be the subject of patent rights. ISO shall not be held responsible for identifying any or all such patent rights.
Further information about this Standard:
Questions or comments about this standard should be addressed to the ISO TC 215 Secretariat:
Audrey E. Dickerson RN, MS
ISO/TC 215 Health Informatics, Secretary
US TAG for ISO/TC 215 Health Informatics, Administrator
Staff Liaison for Nursing Sub-committee to IHE-Patient Care Coordination Domain
Manager, Standard Initiatives
HIMSS
230 East Ohio Street, Suite 500
Chicago, Illinois 60611
TEL: +01-1-312-915-9233
Mobile: +01-1-630-544-0378
FAX: +01-1-312-915-9511
adickerson@himss.org
www.himss.org
Copyright ISO/HL7 2010 " All rights reserved
Introduction
It has been noted that many countries have a strong need to exchange product safety information between a variety of stakeholders in the healthcare domain. Currently many regulatory agencies collect individual case safety reports of adverse events, infections and incidents from consumers, pharmaceutical companies and healthcare professionals. This standard consolidates content and messaging requirements based upon: 1) ISO New Work Item Proposal N545: Health Informatics - Pharmacovigilance - Structure and Data Elements of Individual Case Safety Report (reclassified as ISO 27953), 2) HL7 ICSR Release 1 Normative Standard, and 3) HL7 ICSR Release 2 Draft Standard for Trial Use (DSTU). This release of the standard focuses on the areas of overlap that specifically address the content across the work items.
Readers should note that because of the significant differences in use cases and content requirements across these work items, this standard is published as a multi-part standard. Part 1 of the standard (ISO 27953-1) has been specifically designed to address areas of overlap across the work items and form a messaging framework reference. This framework can be applied to support data exchange requirements described in the many different use cases presented as storyboards in the standard. This framework allows for future development work to be carried out so that additional use cases that are not addressed in this release can be added as new parts to this standard in the future.
Part 2 of this standard (ISO 27953-2) has been created for implementers interested in regulatory reporting for human pharmaceuticals, the initial work has been based upon the International Conference on Harmonisation's (ICH) Revised Guideline E2B(R3): Data Elements for Transmission of Individual Case Safety Reports (version 3.96), November 2008. These initial requirements have been further extended to take into account additional international requirements. This part of the standard is dependent upon the related ISO vocabulary harmonization work items: Data Elements and Structures for the Exchange of Regulated Product Information for Drug Dictionaries (see ISO 11615, 11616, 11238, 11239 and 11240) and Structures and Controlled Vocabularies for Laboratory Test Units for the Reporting of Laboratory Results (see ISO 11595).
Ballot Considerations
This draft standard document is harmonized between ISO, CEN and HL7. It is jointly balloted in all three organizations. In ISO/TC 215 and CEN/TC 251, this is currently a Draft International Standard (DIS). Readers will notice that this document is formatted differently from other HL7 V3 specifications because of the need for consistency with ISO/TC 215 and CEN/TC 251 publications. Ballot reviewers should focus on the content and not submit comments related to the publication format. Ballot comments should be submitted in accordance with the appropriate SDO ballot governing rules, i.e., through ISO National Member Body (NMB) representative, CEN National Member Body (NMB) representative, or through appropriate HL7 membership voting. To help reduce duplication or conflicts in ballot responses among SDOs, HL7 International Affiliates are encouraged to coordinate with their ISO/CEN NMB representatives for submitting comments to this document. The sponsoring ISO, CEN and HL7 committees will jointly consolidate and reconcile all comments per ISO/CEN Vienna Agreement and the ISO/HL7 pilot agreement requirements.
HL7 Version 3 Contents
The format of the document follows the publication standards used by ISO and the standard publishing format for HL7 Version 3 (V3) standards. This hybrid format makes it possible to present the ICSR Refined Message Information Models (RMIMs) and schema files in a seamless way, and to produce material that can be consistently balloted in the CEN, ISO, and HL7 environments. It should be noted that the content for HL7 V3 Messaging Infrastructure (Transmission and Control Act) are included as separate annexes for this ballot to demonstrate support for ISO requirements related to message attachments, batch submissions and acknowledgements. Note that this material is provided as reference only and is not subject to changes or comment as part of the ICSR ballot. Any comments received for this content will be forwarded to HL7 for future consideration.
Within the HL7 publications (which includes the Normative Editions of the standard as well as ballot packages), ICSR material is found as a topic within the Public Health domain. The content presented here includes the skeleton of that domain, with its other topics removed since they are not relevant to the ICSR. We have endeavored to ensure that all hyperlinks are supported. In addition, implementers should note the the standard message schemas will be found associated with interactions and hierarchical message descriptions in the Message Specifications section of this document.
SDO Harmonization
The Joint Initiative on SDO Global Health Informatics Standardization has been formed to enable common, timely health informatics standards by addressing and resolving issues of gaps, overlaps, and counterproductive standardization efforts. The Joint Initiative Charter provides the basis, purpose and structure of the Joint Initiative on SDO Global Health Informatics Standardization. The Individual Case Safety Report (ICSR) Standard was approved as a Joint Initiative project February 2008. The ICSR standard is a candidate for SDO harmonization because of the global interest to improve patient safety by the electronic exchange of high quality, unambiguous, structured data that will support regulatory and patient safety requirements and efficient safety signal detection for patient protection. This standard is generally referred to as the ICSR.
The standard's message specification is based upon the current release of HL7's Reference Information Model, including its data types. The sponsoring committees are aware of the current Joint Initiative Project, ISO/FDIS 21090 Health Informatics -- Harmonized Data Types for Information Interchange (commonly referred in HL7 as Release 2 Data Types.), and the standard will be updated in future releases to incorporate these data types once appropriate technical tools are available to facilitate conversion.
Annexes
Annex A contains information on the HL7 Transmission Infrastructure.
Annex B contains information on the HL7 Control Act Infrastructure.
Annex C contains information on the HL7Version 3 Guide.
Annex D contains information on the HL7 Reference Information Model (RIM)
Annex E contains information on the HL7 abstract datatype specifications
Annex F contains information on the HL7 vocabulary specifications
Annex G contains the ICH Guideline E2B(R3), version 3.96 November 2008 for regulatory human pharmaceutical reporting
1 Scope
1.1 ISO 27593-2: International Regulatory Reporting for Human Pharmaceuticals
This conformance profile for ISO 27593-1 seeks to create a standardized framework for international regulatory reporting and information sharing by providing a common set of data elements and messaging format for transmission of ICSRs for adverse drug reactions (ADR), adverse events (AE), infections, and incidents that may occur upon the administration of one or more human pharmaceutical products to a patient, regardless of source and destination. The standard provides a structure where reports can be exchanged in a clear and unambiguous manner such that the nature of the case, the circumstances in which it arose, and particularly the identity of the medicinal product(s) in question, can be communicated with certainty. Requirements for this use case were initially based upon ICH and conformance for Part 2 includes parallel adoption of ISO vocabulary work items: Data Elements and Structures for the Exchange of Regulated Product Information for Drug Dictionaries (See prEN ISO 11615, 11616, 11238, 11239, and 11240) and Structures and Controlled Vocabularies for Laboratory Test Units for the Reporting of Laboratory Results (See prEN ISO 11595).
1.2 Topics outside ISO 27953-2
This standard does not govern the reporting of ADRs or AE in animals, human exposure to products intended for veterinary use, safety reports related to blood transfusion, medical devices and combination products. Implementers of this standard should note that some combination products (refered to as Advanced Therapies in the EU) contain combinations of pharmaceuticals (which may in some instances be in the form of cells and tissues) along with medical devices or active implantable medical devices. In order to support regions that consider these products to be human pharmaceutical products, the message specifications for Part 2 of this standard (ISO 27953-2) uses an HL7 Common Message Element (CMET) derived from the HL7 Common Product Model to allow the capture of information for the medical device component.
This standard does not govern reporting for foods, dietary supplements, in-vitro diagnostics, homeopathic products, minerals, cosmetics, photo-therapy, radiotherapy and other therapies without products classified as medicinal products. For these use cases and any additional ones please refer to Part 1 of the standard (ISO 27953-1). Finally, some regions may elect to adopt a broader set of data elements based upon Part 1, especially related to clinical trial adverse event reporting. In these cases, implementers should refer to Part 1 to review the available data elements not covered in Part 2's scope.
2 Normative referencesThe following referenced documents are indispensable for the application of this document. For dated references, only the edition cited applies. For undated references, the latest edition of the referenced document (including any amendments) applies.
HL7 2007/2008 Normative Edition: Safety Reporting Topic: Individual Case Safety Report Release 2 Draft Standard for Trial Use
ISO/HL7 21731:2006 Health Informatics -- HL7 version 3 -- Reference Information Model -- Release 1 2006-08-03
ISO/TS 22224:2007, Health Informatics - Electronic Reporting of Adverse Drug Reaction
ISO/FDIS 21090:Health Informatics -- Harmonized Data Types for Information Interchange
3 Terms and DefinitionsNote that there are many different terms used to describe basic concepts in healthcare for different purposes available from ISO, CEN, HL7 and various national and international organisations. Therefore, this standard does not attempt to force adoption of the terms and definitions described in this document. It is intended to be used in combination with any national/regional requirements and, in case of conflict, national/regional requirements will prevail. This being said, for the purposes of this document, the following terms and definitions apply to facilitate conformance and interoperability testing for the regulatory reporting use case for human pharmaceuticals. The terms and definitions included below, are those which do not appear in the HL7 glossary - for which a link is provided. (It is intended to create a single glossary by moving these terms into the HL7 glossary after discussion with the relevant technical committees and working groups)
Detailed terms and definitions relevant to the HL7 messaging specification are provided in the Hierarchical Message Definition (HMD) file and other related sections in HL7's V3 Message Specifications.
Additional Terms and Definitions
The included terms have been identified for their relevance to Human pharmacovigilance and product problem reporting, and which may not appear in the HL Glossary.
3.1 adverse event/adverse experience
Any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have to have a causal relationship with ths treatment.
[ WHO; 2002;]
3.2 acknowledgement message (ICSRACK)
The acknowledgement message is an EDI Message with the information on the result of the acknowledgement of receipt procedure to acknowledge the receipt of one safety message and the safety report(s) contained in the safety file.
[ EMEA; 2002;]
3.3 adverse drug reaction (ADR) or adverse reaction
A response to a drug which is noxious and unintended, and which occurs at doses normally used in man for the prophylaxis, diagnosis, or therapy of disease, or for the modification of physiological function.
[ WHO; 2002;]
3.4 case
A problem requiring investigation, and includes problems that may or may not involve individual or groups of investigative subjects.
3.5 counterfeit medicine
A medicine which is deliberately and fraudulently mislabelled with respect to identity and/or source. Counterfeiting can apply to both branded and generic products and counterfeit products may include products with the correct ingredients or with the wrong ingredients, without active ingredients, with insufficient active ingredients or with fake packaging.
[ WHO]
3.6 drug
(See Medicinal Product)
3.7The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH)
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) is a project that brings together the regulatory authorities of Europe, Japan and the United States and experts from the pharmaceutical industry in the three regions to discuss scientific and technical aspects of product registration. The Observers are WHO, EFTA, and Canada (represented by Health Canada).
ICH]
3.8 healthcare professional
Person entrusted with the direct or indirect provision of defined healthcare services to a subject of care or a population of subjects of care.
EXAMPLE Qualified medical practitioner, pharmacist, nurse, social worker, radiographer, medical secretary or clerk
[ENV 1613:1995] [ISO 21574-7]
3.9 Individual Case Safety Report (ICSR)
It is a document containing the most complete information provided by a reporter at certain point of time to describe suspected adverse reaction(s)/event(s) related to the administration of one or more medicinal product(s) to a subject or a group of subjects.
3.10 interim reporter
A professional or public organization who is monitoring, receiving and assessing ADR reports from health professionals and consumers and reporting significant ADRs to regulatory or statutory authority in its own region.
[ISO/TS 22224:2007]
3.11 marketing authorization holder
An organization, usually a biopharmaceutical firm,that holds a valid marketing authorisation for a medicinal product delivered by the Health Authority of a country.
3.12 Medical Dictionary for Regulatory Activities
Medical Dictionary for Regulatory Activities (MedDRA) terminology for adverse event reporting used globally by the biopharmaceutical industry and regulators to promote consistent reporting and data analysis
3.13 medicinal product
(a) Any substance or combination of substances presented as having properties for treating or preventing disease in human beings;
(b) Any substance or combination of substances which may be used in or administered to human beings either with a view to restoring, correcting or modifying physiological functions by exerting a pharmacological, immunological or metabolic action, or to making a medical diagnosis.
[ISO 11615]
3.14 national pharmacovigilance centre
A single, governmentally recognized centre (or integrated system) within a country with the clinical and scientific expertise to collect, collate, analyse and give advice on all information related to drug safety.
3.15 non-proprietary drug (generic) name
Drug name that is not protected by a trademark, usually descriptive of its chemical structure; sometimes called a public name. International Nonproprietary Names (INN) allocated by WHO, identify pharmaceutical substances or active pharmaceutical ingredients. Each INN is a unique name that is globally recognized and is public property. A nonproprietary name is also known as a generic name.In the US, most generic drug names are assigned by the US Adopted Name Council (USAN).
3.16 pharmacovigilance
The science of activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug-related problem.
[ WHO; 2002;]
3.17 product manufacturer
The organization which is responsible for the manufacture of a medicinal product. This entity may or may not be the same as the marketing authorization holder of the medicinal product.
3.18 regulatory agency (or authorities)
Geopolitical entities have established agencies/authority responsible for regulating products used in health care. The agencies are collectively referred to as regulatory agencies.
3.19 Reference Information Model (RIM)
The HL7 information model from which all other information models, e.g., RMIMS, and messages are derived.
3.20 Refined Message Information Model (RMIM)
An information structure that represents the requirements for a set of messages.
3.21reporter
The primary source of the information, i.e., a person who initially reports the facts provided in the ICSR. This should be distinguished from the sender of the message, though the reporter could also be a sender.
3.22 serious adverse reaction or serious adverse drug reaction
An adverse reaction which is fatal (results in death), is life threatening, requires hospitalization or prolongation of a hospitalization, results in persistent or significant disability/incapacity, results in a congenital anomaly/birth defect or is a medically important condition.
3.23 sponsor
An individual, company, institution or organization which takes responsibility for the initiation, management and/or financing of a clinical trial.
[Article 2e; European Directive 2001/20/EC]
3.24 spontaneous reporting
System whereby case reports of adverse drug reactions/events are voluntarily submitted by any reporters including eg, consumers, health professionals and pharmaceutical manufacturers or marketing authorization holders to the national regulatory authority.
[ WHO; 2002;]
3.25 standard
A technical specification which addresses a business requirement, has been implemented in viable commercial products, and, to the extent practical, complies with recognized standards organizations such as ISO.
3.26 use case
A description of a system's behaviour as it responds to a request that originates from outside of that system.
4 Abbreviated terms
For the purposes of this document, the following abbreviated terms apply:
ADR: Adverse Drug Reaction
AE: Adverse Event
ISO: International Organization for Standardization
CEN: Comite Europeen de Normalisation (European Committee for Standardization, a federation of 28 national standards bodies that are also ISO member bodies)
EU: European Union
HL7: Health Level 7
HMD:Hierarchical Message Description
ICH: The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
ICSR: Individual Case Safety Report
ITS: Implementable Technology Specification
MedDRA: Medical Dictionary for Regulatory Activities
RIM: Reference Information Model
RMIM: Refined Message Information Model
SDO: Standards Development Organization
SGML: Standardized generalized markup language. An ISO standard for describing structured information in a platform independent manner
SNOMED: The Systematized Nomenclature of Human and Veterinary Medicine
SNOMED-CT: Systematized Nomenclature of Medicine-Clinical Terms
UML: Unified Modelling Language
W3C: World Wide Web Consortium
XML: eXtensible Markup Language
5 Regulated Human Pharmaceuticals Message Specification
Conformance relevant to ISO 27953-2
A message or service interface that presents ICSR for regulatory reporting for human pharmaceuticals of adverse events/reactions that may occur upon the administration of one or more medicinal products to a patient must include all of the information and vocabulary requirements specified as mandatory for this part of the standard. The format for ICSR includes provisions for transmitting all the relevant data elements useful to assess an individual adverse drug reaction or adverse event report. The data elements are sufficiently comprehensive to cover complex reports from most sources, different data sets, and transmission situations or requirements; therefore, information for each and every data element will not be available for every transmission. In many, if not most instances, a substantial number of the data elements will not be known and therefore not included in the transmission. Where it was deemed important, provisions for unknown/not applicable were included (e.g., outcome, route of administration). However, since the transmission is intended to be electronic, it was thought to be unnecessary to include provisions to assign values of unknown for all data elements. Different ways of including the same data have been provided to cope with differing information contents: e.g., age information can be sent as date of birth and date of reaction/event, age at the time of reaction/event, or patient age group according to the available information. It is recommended that organizations develop and publish implementation guides that inform users of the standard on how these data elements should be used
Structured data are strongly recommended and provision for including structured information is supported in this standard. In certain instances, there are provisions for the transmission of some free text items, including a full text case summary narrative. Reference documents relating to the case being reported can be transmitted either within an ICSR message or as a separate message to the receivers. Handling of attachments to be sent as part of an ICSR message is supported using the Document attribute. Attachments to be sent separately to the ICSR message are supported using HL7's Message Transmission Infrastructure Specification.
Essential elements to be providedIn most situations when generating an ICSR it is necessary to identify a patient, an adverse event/reaction, a reporter and a suspected medicinal product in order for the message to be considered valid. The minimum information for the transmission of an ICSR and which data elements should be used to fulfil the need for minimum information should be described in an implementation guide.
Types of Regulatory Reports
There is a strong international commitment to improve patient safety and protect public health which drives regulatory authorities to require pharmaceutical companies and manufacturers to report adverse events related to their products. While countries and/or regions establish their own legislation and implementation guides for reporting, it is common to find the following types of reports in many countries:
- Expedited Spontaneous report
- Periodic Spontaneous report
- Expedited Clinical trial report
- Periodic Clinical trial report
- Observational Study report
- Literature report
- Parent-Child report
- Backlog
Human Pharmaceuticals Message Specifications
The Human Pharmaceuticals Message Specifications is best thought of as a constrained version of ISO 27953-1 (Part 1) of the ICSR. That is, it is a model that contains only those elements that are needed to carry the contents required for regulated human pharmaceutical reporting. In order to maintain the integrity of the HL7 model, additional elements are included: the structural attributes of the HL7 classes that contain the attributes that are mapped to the relevant human pharmaceutical reporting data elements, as well as the classes needed to navigate through the model structure. In other words, it is a strict subset of the ICSR Part 1.
Acknowledgement Message Specifications
For more information on acknowledgement messaging, please refer to HL7's Transmission wrapper domain. (Within the ISO/ballot structure, this appears as Annex A.)
Attachment Message Specifications
For more information on attachments to messages, please refer to HL7's Transmission wrapper domain. (Within the ISO/ballot structure, this appears as Annex A.)
|
||||||
|

For details on the interpretation of this section, see the storyboard discussion in the Version 3 Guide.
The storyboards provided for Part 2 of this standard help to describe the reporting situations where the ICSR message would be used. The storyboards referenced in Part 2 were provided by the European Medicines Agency in support of ISO prEN ISO 27953. The storyboards focus on international human pharmaceutical reporting and are based upon the International Conference on Harmonisation's reporting guideline: Revision of the ICH Guideline on Clinical Safety Data Management: Data Elements for Transmission of Individual Case Safety Reports E2B(R3) Version 3.96 13 November 2008. Note that reporting for combination products is supported in Part 2 and these requirements are based upon criteria described in regional legislation in the EU and US. EU reporting is governed by Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on Advanced Therapy Medicinal Products. The amending Directive 2001/83/EC and Regulation (EC) No 726/2004 came into force on 30 December 2008 (http://ec.europa.eu/enterprise/pharmaceuticals/eudralex/vol-1/reg_2007_1394/reg_2007_1394_en.pdf). In the US, reporting for combination products are under the purview of the FDA Office of Combination Products. Readers should also note that while the standard does support these requirements, the standard does not dictate or enforce that ICSR be specifically used for these products. It is expected that the EU and US regualtory authorities will issue separate guidance about the use of ICSR for these products at a later date and implementers should contact the appropriate regulatory authority prior to using ICSR for these products.
| Human Pharmaceuticals Report Create | |
| HumanPharmaceuticals Report Revise | |
| Human Pharmaceuticals Report Retract |
Note that this storyboard provides an example of the multiple steps or stages of ICSR review and follow up responses. However, the actual steps or stages of adverse event reporting may vary, and the steps described here should not be interpreted as a harmonized process flow for every organization.
The Event
On 4th of February 2007 Mr. Patient, a 42 year old male patient experienced dizziness and heavy chest pain after taking two oral ramapril 75mg tablets. Feeling better after 20 minutes, he phoned the marketing authorization holder Big Pharma to report his experience stating that he believes he might have had a heart attack.
Creation of an initial ICSR report
Big Pharma collected all the details of the report in their pharmacovigilance IT system, and recorded dizziness, chest pain and heart attack as the reported events. These events were flagged as 'not confirmed by health care professional'. As the report originated from a consumer, the report did not qualify for submission in certain regulatory jurisdictions. Therefore Big Pharma submitted the report electronically as an expedited case only to the appropriate drug regulatory authorities requiring consumer reports.
Interaction: IHuman Pharmaceuticals Report Create(PORR_IN049016)
Regulatory authority response to the Big Pharma Report - Note that this function is outside the ICSR and relates to HL7 accept acknowledgement interfaction ID (MCCI_IN000002UV01
The electronic report is received by the appropriate regulatory agencies and it is checked against their requirements for a valid expedited report. The report is found to be valid and a positive acknowledgement is returned to Big Pharma to confirm receipt and acceptance of the expedited report.
Marketing Authorisation Holder receives follow-up information from physician
Big Pharma requested permission from Mr. Patient to consult his physician, Dr. Care. The physician informs Big Pharma that Mr. Patient had come to see him and states that the patient did not experience a heart attack but instead has been diagnosed with angina.
Creation of a Revised (follow up) ICSR
Big Pharma complets an updated ICSR using their own IT system. The reported events where 'angina' (flagged as 'confirmed by health care professional'), 'dizziness' (flagged as 'not confirmed by health care professional'), 'chest pain' (flagged as 'not confirmed by health care professional') and 'heart attack' (flagged as 'not confirmed by health care professional') were included in the revised ICSR. Big Pharma submitted the updated report electronically to the drug regulatory authorities that had received the initial ICSR. Since the revised report contains information that has been confirmed by a health care professional, the report now qualifies for submission to other drug regulatory authorities as well and Big Pharma sends the report electronically to the other drug regulatory authorities requiring medically confirmed reports.
Interaction: Human Pharmaceuticals Report Create(PORR_IN049017)
Regulatory Authority response - Note that this function is outside the ICSR and relates to HL7 accept acknowledgement interaction ID (MCCI_IN000002UV01
The electronic ICSR is received by the drug regulatory agencies and it is checked against their requirements for a valid expedited report. The report is found to be valid and a positive acknowledgement (outside of the ICSR reporting standard) is returned to Big Pharma to confirm receipt and acceptance of the expedited report. For scientific analyses the drug regulatory agencies can distinguish between events reported by consumers and events reported by health care professionals.
The Event
On 4th of June, 2007, Dr. A. Cure, contacted Nobel X company by telephone to see if the company's product Atrovim and antihypertensive drug could of caused syncope in a 53 year old male patient. The drug information officer at the company confirmed that Syncope was listed in the company's product information as a possible side effect of the medicine. The doctor provided no further information about the case at the time
Creation of the ICSR Nullification Report
The drug information officer at Nobel Company records the details of the telephone conversation into the company's pharmacovigilance system and details of the adverse drug event are sent to drug regulatory authority. Nobel Company also writes to Dr A Jones including a questionnaire to find out further details about the event.
Interaction: Human Pharmaceuticals Report Create(PORR_IN049016)
Nullification of the Report
Dr A Jones, writes back to Noble Company on the 7th of July and informs the company that his patient did not in fact receive Atrovim and the syncope was not related to any medication taken by the patient.
A Nobel Company representative updates the case report on the company database and marks the report as deleted. A nullification report is created by Nobel giving the nullification reason that "Further information received from the reporting physician states that the patient did not receive the suspect drug and the event was unrelated to any medication taken by the patient". The nullification report is then sent to the regulatory authority. The regulatory authority on receipt of the nullification marks the report within their system as "Nullified" and no longer uses the case for scientific evaluation but the case is kept for audit purposes.
Interaction: Human Pharmaceuticals Report ?Withdraw(PORR_IN049018)
The event
A 22 year old woman ordered zolpidem (indicated for short-term treatment of insomnia), via an internet pharmacist. After taking two tablets she experienced difficulty in breathing, muscle spasms and muscle stiffness and emergency medical treatment in the General Hospital X in Manchester was necessary. The patient brought the medication to the hospital and the treating physician Dr Caregiver informed the marketing authorisation holder shown on the package (Big Pharma) of these events. On request of Big Pharma Dr. Caregiver sent the package with the remaining zolpidem tablets to Big Pharma.
Creation of the Initial ICSR Counterfeit Medicine Report
The drug information officer at Big Pharma records the details of the telephone conversation with Dr. Caregiver into the company's pharmacovigilance system and details of the adverse drug event (with zolpidem stated as suspected medication) are sent to drug regulatory authority.
Interaction: Human Pharmaceuticals Report Create(PORR_IN049016)
Authority response - Note this is outside of the ICSR standard and refers to HL7 accept acknowledgement interfaction ID (MCCI_IN000002UV01)
The electronic report is received by the agency from Big Pharma and it was checked against their requirements for a valid report. The report is found to be valid and a positive acknowledgement is returned to Big Pharma to confirm receipt and acceptance of the report.
Marketing authorisation holder receives results of analysis
Laboratory analysis conducted by Big Pharma confirmed that the tablets contained haloperidol (an antipsychotic drug known to cause symptoms as difficulty in breathing, muscle spasms and muscle stiffness) instead of the intended drug zolpidem.
Creation of an ICSR Follow-up Report
Big Pharma updates the record with this new information and submits a follow-up ICSR electronically to the drug regulatory authority. The suspected medication reflected in the ICSR was retained as zolpidem, but this was flagged as 'counterfeit' in the regulatory database.
Interaction: Human Pharmaceuticals Report Revise(PORR_IN049017)
Authority response (outside the ICSR standard and refers to HL7 accept acknowledgement interaction ID (MCCI_IN000002UV01)
The electronic report is received by drug regulatory authority and it was checked against their requirements for a valid report. The report is found to be valid and a positive acknowledgement is returned to Big Pharma to confirm receipt and acceptance of the report. In view of the public health importance the drug regulatory authority decides to further investigate this issue.
The Event
A patient was enrolled in a UK based multi-centre observational study (Protocol No 012413/01) looking at the prevention of Osteoporosis using Hormone Replacement Therapy in post-menopausal women.
The patient has been taking unopposed oral Oestrogen 625g since June 1999 as she had a full hysterectomy in November 1998 due to endometriosis. The patient was enrolled in the study in April 2000 and was being monitored every 6 months at the Manchester general hospital. In May 2004 the patient was diagnosed with breast cancer during a routine mammogram screening. The patient has been booked for surgery and has since stopped taking Oestrogen. The patient is also taking Bendrofluazide Oral for Hypertension and medical history of smoking and being overweight.
Creation of Observational Study ICSR Report
The physician responsible for the patient (Dr A Jones at the Manchester general hospital, Manchester UK) informed SME Company of the event in their study and they in turn created an electronic report to submit to the concerned regulatory authority using their own IT system. The company provides the following assessment of the case: It is possible that the investigational product caused this reaction.
Interaction: Human Pharmaceuticals Report Create(PORR_IN049016)
Regulatory Authority response (note that this acknowledgement message is outside the ICSR standard and refers to HL7 accept acknowledgement interaction ID (MCCI_IN000002UV01)
The electronic report is received by an agency from SME company and it is checked against their requirements for a valid study report. The report is found to be valid and a positive acknowledgement is returned to SME Company to confirm receipt and acceptance of the report.
The Event
In the UK a multi-centre clinical trial with the EU Clinical trial authorisation number (EUDRACT No. 2004-09102-03) is being conducted to evaluate the efficacy and tolerability of Danthium, a new substance, in post-menopausal women with breast cancer hormone receptor positive tumours. The sponsor of the trial assigned the protocol No 0105798/01.
A 50 year-old female patient was enrolled in this trial with the patient ID 125-0871 and one week after the third cycle of chemotherapy treatment with intravenous Danthium 20 mg/kg once a week, the patient developed a fever (38o C) and diarrhoea (11th May 2003). The patient was hospitalised. Blood tests were performed and the patient was discovered to have neutropenia. This adverse event was considered as serious and unexpected. Specific lab test information such as the lab test date, name, results (including structured units of measurement) and an indication of whether or not the lab test was within the normal range were included in the ICSR report. The patient was treated with G-CSF and recovered a week later. The patient is a smoker and has a family history of breast cancer. The patient was also concomitantly taking oral domeridone for Nausea.
Creation of the Clinical Trial Initial Report
The investigator responsible for the patient (Dr. Davis at Cardiff University hospital, Cardiff, Wales) informed the sponsor (Big Company) of the event and the sponsor in turn created an electronic report to submit to the appropriate regulatory authorities using their own IT system.
The company also provides their causality assessment of the case, which reflects that it is likely that the investigational medicinal product caused the reaction.
Interaction: Human Pharmaceuticals Report Create(PORR_IN049017)
Regulatory Authority response (outside of the ICSR functionality and relates to HL7 accept acknowledgement interaction ID (MCCI_IN000002UV01)
The electronic report is received by an agency from Big Company and it is checked against their requirements for a valid report. The report is found to be valid and a positive acknowledgement is returned to Big Company to confirm receipt and acceptance of the report.
The Event
In the EU a multi-state clinical trial with the EU Clinical trial authorization number (EUDRACT No. 2004-09102-03) is being conducted in Germany and Netherlands to evaluate the efficacy and tolerability of Danthium, a new substance, in post-menopausal women with breast cancer hormone receptor positive tumors. The sponsor of the trial assigned the protocol No 0105798/01. The clinical trial authorization number assigned by the German authorities is '12345678' and the authorization number assigned by the Dutch authorities is 'NL12345.001.07'
A 50 year-old female patient with the patient ID 125-0871 was enrolled in this trial in a German centre and one week after the third cycle of chemotherapy treatment with intravenous Danthium 20 mg/kg once a week, the patient developed a fever (38o C) and diarrhea (11th May 2003). The patient was hospitalized. Blood tests were performed and the patient was discovered to have neutropenia. This adverse event was considered as serious and unexpected.
Lab Test Results:
Date________Test Name___________Test Result___Test Units____Normal Range
12/05/2003___Neutrophil Count______4.0 x10e9_____cells/l_______2.0 - 7.5
12/05/2003___WBC_______________6.8 x10e9_____cells/l_______4.0 - 11.0
19/05/2003___Neutrophil Count______0.8 x10e9_____cells/l_______2.0 - 7.5
19/05/2003___WBC_______________4.5 x10e9_____cells/l_______4.0 - 11.0
26/05/2003___Neutrophil Count______3.8 x10e9_____cells/l_______2.0 - 7.5
26/05/2003___WBC_______________6.3 x10e9_____cells/l_______4.0 - 11.0
The patient was treated with G-CSF and recovered a week later. The patient is a smoker and has a family history of breast cancer. The patient was also concomitantly taking oral domeridone for Nausea.
Creation of Clinical Trial Report
The investigator responsible for the patient (Dr B. Bernard at Berlin University hospital, Berlin) informed the sponsor (Big Company) of the event and the sponsor in turn created an electronic report to submit to the concerned regulatory authorities using their own IT system. The company provides the following assessment of the case: It is likely that the investigational medicinal product caused this reaction.
Interaction: Human Pharmaceuticals Report Create(PORR_IN049016)
Regulatory Authority response - This function is outside ICSR and relates to HL7 accept acknowledgement interaction ID (MCCI_IN000002UV01)
The electronic report is received by an agency from Big Company and it is checked against their requirements for a valid report. The report is found to be valid and a positive acknowledgement is returned to Big Company to confirm receipt and acceptance of the report.
The Event
The event is a literature report of a 58 year old male UK patient who had anaphylaxis after 2 days of taking oral captopril tablets (10mg twice a day) for Hypertension in combination with oral aspirin capsules (75mg once a day) for stroke prophylaxis. The patient was then admitted to hospital due to an anaphylactic reaction. The patient then stopped taking the captopril and was treated with prednisolone and subsequently recovered. The event was considered by the author to be life threatening
The author of the article suspects that there was interaction between the captopril and the aspirin that led to anaphylaxis, which presented initially as shortness of breath and severe hypotension.
The patient was taking captopril from the 15th April 1999 until the 17th April 1999 and was taking aspirin from December 1996 and is continuing to take it.
The patient has a history of diabetes and a cerebrovascular accident in 1996. The patient is also concurrently taking Metformin and Insulin for his diabetes. The patient has also taken Ibuprofen in May 1997 for a headache, the patient then experienced an allergic rash that was attributed at that time to the Ibuprofen, and the patient has not used Ibuprofen since then.
The author is based in the UK and the report was published in an American Journal, reference below:
Ahmed A et al. Interaction between aspirin and angiotensin-converting enzyme inhibitors J American Geriatric Soc 2003; 49(6): 1291-2
Creation of the ICSR Literature Report
The Marketing Licence holder Nobel Company regular checks the worldwide literature for case reports appearing for its products. This article is subsequently found and this report has to be submitted to the UK Medicines Agency (MHRA). Using their own IT system they create and send the report electronically to the MHRA. The company assigned the following unique case identifier to the case report GB-Nobel-Testcase04.
Interaction: Human Pharmaceuticals Report Create(PORR_IN049016)
MHRA response: Note that this positive acknowledgement message is handled outside the ICSR functionality and refers to HL7 accept acknowledgement interaction ID (MCCI_IN000002UV01)
The electronic report is received by the agency and it was checked against their requirements for a valid report. The report is found to be correct and a positive acknowledgement is returned to Nobel Company to confirm receipt and acceptance of the report.
This storyboard was created to help illustrate how the ICSR can be used for submitting electronic medicine-related events that were originally sent using a paper-based process.
The Event
On 4th of June, 1996, Mr A. Past, a 62 year old male was admitted to Berlin General Hospital X with anaemia. The patient subsequently recovered by 6th August 1996.
The patient had been taking oral Ibuprofen 400mg Tablets twice a day, for arthritic pain of the hands. The patient had not been taking any concomitant medications at the time.
Creation othe ICSR Backlog Report
The reporting physician Dr H. Provider contacted the marketing authorisation holder (Big Company) initially on the 17th June 1996 to inform them of this case. The physician then subsequently contacted the company again on the 7th August 1996 to inform them about the recover of the patient.
The company collected all the details of the event within their own records system. The report was then submitted to the local regulatory agency at that time on paper as was required in 1996. In 2006 the company was requested by the regulatory agency to resubmit this case in electronic format as required in regional requirements. The report is created by the company in electronic format (ICSR) with the message transmission type selected as backlog so that the report is clearly identifiable.
Interaction: Individual Case Safety Report Create(PORR_IN049016)
Regulatory Agency response (outside of the ICSR standard functionality)The electronic report is received by the agency from Big company and it was checked against their requirements for a valid backlog report. The report is found to be valid and a positive acknowledgement is returned to Big Company to confirm receipt and acceptance of the backlog report.
|
||||||||
|
For details on the interpretation of this section, see the discussion of application roles and their relationships in the Version 3 Guide.
Indicates the party that receives notification of the human pharmaceuticals related adverse.
Indicates the party that provides notification of the human pharmaceuticals related adverse event.
|
||||||||||
|
For details on the interpretation of this section, see the discussion of trigger events in the Version 3 Guide.
| Type: |
Indicates that a prior report containing notification of an eligible case associated with an adverse event related to the use of a human pharmaceutical is being revised or amended. The revision may include the communication of attachments relevant to the case.
| Type: |
Indicates that a report containing notification of an eligible case associated with the use of a human pharmaceutical is ready for transmission to an eligible receiver. The notification includes the report of an adverse event or reaction.
| Type: |
Indicates that a prior report containing notification of an eligible case associated with the use of a human pharmaceutical is being withdrawn and should be nullified
|
||||||||
|
For details on the interpretation of this section, see the description of RMIMs in the Version 3 Guide.

| Parent: | Public Health Reporting (PORR_DM000000UV) |
Model Overview
The Human Pharmaceuticals Base Model RMIM is designed to support a report about an investigation into an adverse event(s) or reaction(s) suffered by a person that experienced an intervention (substance administration or procedure) within a therapeutic context. The suspect event may or may not have a causal relationship with the intervention and the model supports events suffered by associated persons e.g., mother/child or siblings.
The RMIM is oriented around the following concepts:
- Investigation: Information concerning the activity carried out to investigate and understand an adverse event. This is the focal class for the model. Note that participants (e.g., reporters, senders and receivers) in the investigation, as well as documents and/or observations related to the investigation may be recorded.
- Control Act: Information concerning the reporting of the investigation and/or related investigations (reports) for which information may be included. Note that information for the author and/or primary recipient of the report may be included in this structure
- Adverse Event Assessment: An ICSR report is based on the assessment that an adverse event has occurred. The assessment is linked to the person affected by an adverse event and to the suspect product that may have caused the event. An investigation may or may not be linked to a causality assessment. Note that the model supports additional observations related to the assessment, e.g., seriousness, severity or toxicity ratings for adverse events.
- Causality Assessment: Judgements concerning the cause or causes of an adverse event. Includes the authors, methods, results and conclusions
- Primary Role/Investigation Related Entity: For an adverse event assessment, the role code provides information about the specific role a person plays, e.g., a patient or research study participant, which suffered the adverse event. Note that relevant information about the person and the event is captured in the related Human Pharmaceuticals Product Reporting Relevant Information CMET. In addition, if the person is enrolled in a clinical trial, the trial can be identified as well.
- Study Enrollment: Relevant information about the clinical trial or study that the subject may be involved in.
- Related Investigation: Information about other reports (primary source reports, prior submissions or secondary case transmissions) linked to the investigation. This can include reports of the same adverse event based on investigations carried out by other parties, or reports of adverse events that are considered to be related, e.g., mother/child reports.
- Assigned Entity: Information for an entity, whether a person or organization that is involved in the reporting or assessment of the adverse event or product problem.
- Product Information: Readers should note that product information is captured using a CMET (R_ProductReportable COCT_RM630000UV) derived from the Common Product Model (CPM). This CMET may adjust over time as updates are made to the CPM. The ISO and HL7 committees will continue work to harmonize requirements for the ISO Identification of Medicinal Products work items. The data elements reflected in this release of CPM DSTU Release 1 support the current pharmacovigilance requirements of ISO 27953-1 and ISO 27953-2. Readers should note that ISO 27953 will use the most current version of CPM in its final international standard publication set for early 2011. Navigation to the CMET content is through the Human Pharmaceuticals base model Investigation Component Choice box and the consumable participation in the A_HumanPharmaceuticalsPRRI CMET. The model captures information about products used by the patient or an associated entity. This includes products that are implicated or considered suspects (primary) that may have an association with the adverse event. It also includes products that are (delete or) used around the same time (usually referred to as concomitant or interacting products). This model captures information about associated product acts such as production, testing, supply, and distribution, as well as information about the product itself e.g., dosage form, ingredients, lot number, etc.
This message, as with all HL7 V3 specifications, includes a Control Act structure which contains information about the actual report transmission. (The reader should refer to the HL7 V3 Infrastructure Management Domain for more details on this structure.)
Please refer to the HMD documentation for more detailed information on classes and attributes.
| HumanPharmaceuticalsBaseRMIM | PORR_HD049016UV01 |

| Parent: | Public Health Reporting (PORR_DM000000UV) |
Model Overview
The A_HumanPharmaceuticalsPRRI RMIM captures information about the acts that describe how a product was used by an investigative subject. The information includes use of a product (substance administration and device procedures) and any associated clinical or laboratory information directly related to the product's use at a particular point in time, e.g., related to an adverse event, or as part of a subject's medical history. The model also supports other patient care or healthcare-related processes such as actions taken to mitigate or reduce harm. A choice box structure is used (based upon HL7's A_SupportingClinicalInformation CMET) to capture the relevant information needed to evaluate the case. It is expected that implementers will need to provide the appropriate code sets needed to distinguish between the various kinds of acts or observations used in the CMET. Conformance for Part 2 uses the Medical Dictionary for Regulatory Activities (MedDRA) coding and other codes provided by ICH. Examples include Organizer codes to group medical or medications history, Observation codes for patient specific observations such as age and weight, and clinical diagnosis or conditions such as Diabetes.
The information model supports:
- Drug administration and device procedure information: date, time, quantity, dose, and route of administration (or target site). Note dose form is captured as product information in the R_ProductReportable CMET (COCT_RM630000UV).
- Component acts or concomitant therapies.
- Authors and/or performers of healthcare acts or services.
- Specimen collection and lab testing.
- Organizer class to facilitate grouping of related acts relevant to a point in time, e.g., vaccines given within the last 4 weeks prior to an adverse event or medical history.
Please refer to the HMD documentation for more detailed information on classes and attributes.
| HumanPharmaceuticalsPRRIModel | PORR_HD049023UV01 |
|
||||||||
|
For details on the interpretation of this section, see the description of HMDs in the Version 3 Guide.
This HMD includes the data attributes that may be needed to file an ICSR.
| A_HumanPharmaceuticalsPRRIModelUniversal | PORR_MT049023UV |
| HumanPharmaceuticalsBaseMT | PORR_MT049016UV01 |
The A_HumanPharmaceuticalsPRRI HMD contains the serialized elements from the corresponding RMIM.
| R_ProductReportableUniversal | POCP_MT020200UV01 |
| HumanPharmaceuticalsPRRIMT | PORR_MT049023UV01 |
|
||||||||||
|
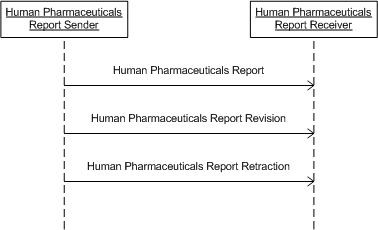
For details on the interpretation of this section, see the definition of Interactions in the Version 3 Guide.
The interaction supports revisions or amendments to previously sent individual case safety report messages.
| Trigger Event | Human Pharmaceutical Revise Notification | PORR_TE049017UV01 |
| Transmission Wrapper | Send Message Payload | MCCI_MT000100UV01 |
| Control Act Wrapper | Trigger Event Control Act | MCAI_MT700201UV01 |
| Message Type | HumanPharmaceuticalsBaseMT | PORR_MT049016UV01 |
| Sender | Human Pharmaceuticals Report Sender | PORR_AR040011UV01 |
| Receiver | Human Pharmaceuticals Report Receiver | PORR_AR040012UV01 |
The interaction supports the communication of a new individual case safety report. Note that this interaction supports both initial and follow up reports.
| Trigger Event | Human Pharmacetical Complete Notification | PORR_TE049016UV01 |
| Transmission Wrapper | Send Message Payload | MCCI_MT000100UV01 |
| Control Act Wrapper | Trigger Event Control Act | MCAI_MT700201UV01 |
| Message Type | HumanPharmaceuticalsBaseMT | PORR_MT049016UV01 |
| Sender | Human Pharmaceuticals Report Sender | PORR_AR040011UV01 |
| Receiver | Human Pharmaceuticals Report Receiver | PORR_AR040012UV01 |
The interaction supports the retraction of a previously sent individual case safety report. Note that this is commonly referred to as a nullification report.
| Trigger Event | Human Pharmaceutical Withdraw Notification | PORR_TE049018UV01 |
| Transmission Wrapper | Send Message Payload | MCCI_MT000100UV01 |
| Control Act Wrapper | Trigger Event Control Act | MCAI_MT700201UV01 |
| Message Type | HumanPharmaceuticalsBaseMT | PORR_MT049016UV01 |
| Sender | Human Pharmaceuticals Report Sender | PORR_AR040011UV01 |
| Receiver | Human Pharmaceuticals Report Receiver | PORR_AR040012UV01 |
| Return to top of page |